
Разработка и внедрение программ, направленных на защиту окружающей среды и сохранение флоры и фауны водоемов, проходят в каждом регионе. Предприятия, осуществляющие слив сточных вод в естественные водоемы, несут бремя ответственности, а соблюдение ими правил организации стоков, очистки и обеззараживания загрязненной и бывшей в использовании воды контролируются на правительственном уровне. Именно поэтому вопросу выбора наиболее эффективной, безопасной и надежной системы очистки сточных вод уделяется особое внимание.
Метод коагуляции – новое направление в очищении и обеззараживании сточных вод. Сегодня существует несколько методов очищения загрязненных сточных вод, среди которых особую популярность получил коагуляционный метод очистки воды, относящийся к категории химических, не представляющий угрозы природе. Коагуляция воды направлена на качественную очистку подвергающихся обработке и переработке жидкостей, которые используются на промышленных объектах. Проведенное очищение и обеззараживание загрязненной воды позволяет вторично ее использовать или осуществлять сброс в реки, не нанося вред окружающей среде, живой флоре и фауне, что особенно важно сегодня, когда вопросам экологии и сохранения природных ресурсов уделяется столько внимания. Основная задача системы очистки стоков – удаление загрязнений, для которых подбирается строго определенный коагулянт для очистки воды с целенаправленным действием. Коагуляция позволяет эффективно очистить сточные воды с использованием специальных реагентов, нахождение в воде которых впоследствии не приведет к нарушению микрофлоры природного водоема, куда идет слив сточных вод предприятия.
Важнейшие экспериментальные факты, способные служить базой для построения теории коагуляции базируются на многообразии причин, вызывающих коагуляцию, на коагулирующем действии электролитов, на влиянии величины заряда иона-коагулятора [1–6].
Установленные закономерности коагуляции нашли свое выражение в правиле Шульце-Гарди, высказанном еще в конце XIX века [1–6].
В ходе развития представлений об устойчивости и механизмах коагуляции гидрофобных коллоидов растворами электролитов возникло много теорий, которые пытались объяснить три вопроса [1–5]:
1) почему коагуляция наступает при определенной концентрации электролита-коагулятора;
2) почему при этом основную роль играет концентрация иона, несущего заряд, противоположный заряду частицы;
3) почему влияние заряда иона-коагулятора подчиняется закономерности, выраженной правилом Шульце-Гарди.
Все теории коагуляции в основном подразделяются на адсорбционные и электростатические [1–6]. Современная физическая теория устойчивости и коагуляции коллоидных систем была развита Б.В. Дерягиным и Л.Д. Ландау (1937), Э. Фервеем и Я. Овербеком (1941). В соответствии с первыми буквами фамилий авторов теория носит название ДЛФО. Согласно этой теории, между любыми частицами при их сближении возникает расклинивающее давление разделяющей жидкой прослойки в результате действия сил притяжения и отталкивания. Состояние системы зависит от баланса энергии притяжения и энергии отталкивания. Преобладание энергии отталкивания приводит к устойчивости системы. Преобладание энергии притяжения вызывает нарушение агрегативной устойчивости, т.е. коагуляцию [1, 5, 6].
Коагуляция сложный процесс, который зависит как от природы золя, так и от природы коагулирующих электролитов. Настоящая работа посвящена изучению закономерностей и механизма коагуляции под действием индифферентных электролитов на примере коллоидной системы – золя Fe(OH)3.
Васильевым С.Е. выполнена экспериментальная часть работы и изучен механизм коагуляции.
Основная часть
Коагуляция гидрофобных золей. Факторы, вызывающие коагуляцию
Лиофобные дисперсные системы (золи, суспензии, эмульсии) агрегативно неустойчивы, поскольку обладают избытком поверхностной энергии Гиббса. Процесс укрупнения частиц (коагуляция) протекает самопроизвольно, так как он ведет к уменьшению удельной поверхности и снижению поверхностной энергии Гиббса. Коагуляция представляет собой процесс слипания (или слияния) частиц дисперсной фазы при потере системой агрегативной устойчивости [1–6].
При коагуляции изменяются физико-химические свойства систем: появляется мутность, снижается осмотическое давление, изменяются электрическая проводимость и характер вязкости. На изменении физико-химических свойств основаны методы наблюдения и изучения процесса коагуляции.
Фактором, вызывающим коагуляцию, может быть любой агент, нарушающий агрегативную устойчивость системы, например изменение температуры (сильное нагревание или охлаждение вплоть до замораживания), механическое воздействие (интенсивное встряхивание, перемешивание, перекачивание по трубам), действие света и различного рода излучений, действие электрических зарядов. Однако наиболее важным фактором является действие электролитов. Электролиты, добавляемые к золям, чрезвычайно быстро и резко влияют на толщину ДЭС и на z-потенциал, являющийся одним из главных факторов устойчивости гидрофобных коллоидных систем.
Коагуляция под действием электролитов. Правило Шульце-Гарди
Наблюдения Г. Шульце (1882) показали, что коагулирующей способностью обладает один из ионов добавляемого электролита (ион-коагулятор). Коагулирующая способность иона-коагулятора возрастает с увеличением его заряда (правило Шульце). Несколько позже М.Гарди (1900) нашел, что заряд коагулирующего иона всегда противоположен заряду коллоидной частицы (правило Гарди). Следовательно, коагуляцию отрицательного золя вызывают катионы добавленного электролита. Для золя с положительно заряженными частицами ионами-коагуляторами являются анионы [1–6].
Закономерности, найденные Шульце и Гарди и подтвержденные многочисленными исследователями, известны как правило Шульце-Гарди: коагулирующим действием обладает тот ион электролита, который имеет заряд, противоположный заряду гранулы; коагулирующее действие тем сильнее, чем выше заряд иона-коагулятора (правило значности).
Порогом коагуляции (g) называют наименьшее количество электролита, необходимое для коагуляции 1 м3 золя. Его выражают в ммоль/л или моль/л.
Для расчета порога коагуляции достаточно умножить концентрацию электролита с (кмоль/м3) на объем его раствора, вызвавшего коагуляцию, V (м3), и произведение разделить на взятый объем золя W (м3):
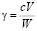 (кмоль/м3). (1)
(кмоль/м3). (1)
(при V<< W).
Б.В. Дерягиным выведено уравнение зависимости порога коагуляции от валентности коагулирующего иона Z:
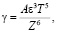 (2)
(2)
где А – обобщенная константа; e – диэлектрическая проницаемость среды; Т – температура.
Величину, обратную порогу коагуляции, называют коагулирующей способностью и обозначают Vк. Коагулирующую способность рассчитывают по формуле
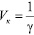 . (3)
. (3)
Коагулирующая способность выражает число объемов золя, скоагулированных 1 моль (или ммоль) иона-коагулятора.
Необходимо иметь в виду, что величина порога коагуляции зависит от ряда условий: от момента его фиксирования после внесения электролита, от метода наблюдения, от концентрации исследуемого золя и др., – которые необходимо указывать при определениях порога коагуляции. Наиболее распространенные методы определения порога коагуляции состоят в наблюдениях за изменением светорассеяния (через определенное время после смешивания золя с электролитом) или в титровании золя раствором электролита до начала явной коагуляции.
Экспериментальная часть
Для изучения процесса коагуляции получаем агрегативно устойчивый золь Fe(OH)3 по реакции гидролиза:
FeCl3(изб.) + H2O = Fe(OH)3 + HCl.
В колбу наливаем 150 мл дистиллированной воды и нагреваем до кипения. В кипящую воду порциями приливаем 15 мл 5%-го раствора хлорного железа и продолжаем кипячение получившегося раствора ещё 7 минут до получения красно-вишнёвого цвета.
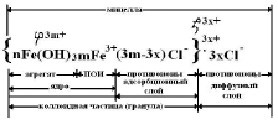
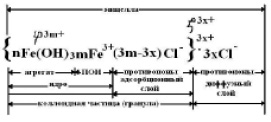
Формула мицеллы золя Fe(OH)3
Для золя Fe(OH)3 с положительно заряженными коллоидными частицами ионами-коагуляторами по правилу Шульце-Гарди во вводимых в золь электролитах: NaCl, Na2SO4, K3[Fe(CN)6] – будут являться анионы: Cl-, SO42–, [Fe(CN)6]3–, т. к. их заряд противоположен заряду коллоидных частиц.
Определение влияния заряда иона-коагулятора на коагуляцию
В три пробирки наливаем 5 мл полученного и охлажденного до комнатной температуры золя Fe(OH)3. В каждую пробирку по каплям добавляем из бюретки раствор электролита до появления первых признаков коагуляции (мутность раствора).
Природа добавляемых электролитов указаны в табл. 1. Полученные результаты сводим в табл. 1.
Таблица 1
|
Наименование электролита |
Концентрация электролита |
Объем электролита, пошедшего на коагуляцию, мл |
Объем электролита, пошедшего на коагуляцию в пересчете на 0,001N раствор, мл |
lgVэл. |
|
NaCl |
3 N |
4 |
1200 |
3,1 |
|
Na2SO4 |
0,01 N |
2 |
0,3 |
|
|
K3[Fe(CN)6] |
0,001 N |
0,5 |
0,5 |
-0,3 |
Строим график, откладывая на оси абсцисс логарифм количества мл 0,001 N раствора электролита, вызвавшего коагуляцию, а на оси ординат величины зарядов ионов коагуляторов.
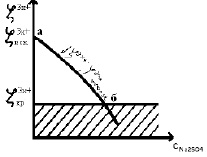
Рис. 1. График зависимости заряда иона-коагулятора от логарифма объема электролита
Из графика делаем вывод: чем больше заряд иона-коагулятора, тем меньше объём электролита необходим, чтобы вызвать коагуляцию.
Определение порога коагуляции электролитов
Приготовили 12 пробирок для исследования коагулирующей способности трех электролитов: NaCl, Na2SO4, K3[Fe(CN)6]. Определение порога коагуляции каждого электролита осуществляли в четырех пробирках. Схема заполнения каждой серии из четырех пробирок представлена в табл. 2.
Таблица 2
|
Номер пробирок |
1 |
2 |
3 |
4 |
|
Золь, мл |
5 |
5 |
5 |
5 |
|
Вода, мл |
4,5 |
4,0 |
3,0 |
1,0 |
|
Объём электролита, вызвавший коагуляцию, мл |
0,5 |
1,0 |
2,0 |
4,0 |
|
Коагуляция через 20 мин |
||||
|
NaCl |
- |
- |
- |
+ |
|
Na2SO4 |
+ |
+ |
+ |
+ |
|
K3[Fe(CN)6] |
- |
+ |
+ |
+ |
После заполнения каждой пробирки всеми составляющими содержимое тщательно перемешивали. Появление или отсутствие коагуляции фиксировали в табл. 2, через 20 минут. Появление коагуляции (+), отсутствие (–).
Порог коагуляции электролитов вычисляли по формуле
g = 100СV;
где С – молярная концентрация электролита; V – минимальный объем электролита, вызвавший коагуляцию.
γNaCl = 100∙3∙4 = 1200 ммоль/м3
γNa2SO4 = 100(0,01/2) ⋅0,5 = 0,25 ммоль/м3
gK3[Fe(CN)6] = 100(0,001/3) ⋅1 = 0,033 ммоль/м3
Коагулирующую способность электролитов рассчитывали по формуле (3)
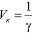 .
.
VNaCl = 1/1200 = 0,83 ∙ 10–3 м3/ммоль;
VNa2SO4 = 1/0,25 = 4 м3/ммоль;
VK3[Fe(CN)6] = 1/0,033 = 30,3 м3/ммоль.
Вывод: чем больше заряд иона-коагулятора, тем меньше у него порог коагуляции, тем выше коагулирующая способность электролита.
Влияние электролитов на j- и z-потенциалы ДЭС
Механизм коагуляции
Согласно теории ДЛФО, при быстрой коагуляции коллоидных систем электролитами возможны два основных механизма [1, 5, 6]:
1) концентрационная коагуляция, при которой наблюдается снижение величины z-потенциала при неизменяющемся j-потенциале поверхности;
2) нейтрализационная коагуляция, происходящая в результате снижения поверхностного j-потенциала частиц и параллельного снижения величины z-потенциала.
Тип коагуляции зависит как от свойств коллоида, так и от природы прибавляемых электролитов.
Распределение ионов в ДЭС, особенно в диффузной части, является функцией концентрации электролитов в объеме раствора.
По отношению к каждому золю электролиты можно подразделить на индифферентные и неиндифферентные. В индифферентных электролитах отсутствуют ионы, которые могли бы избирательно по правилу Фаянса-Панета адсорбироваться на поверхности микрокристаллов. Неиндифферентные электролиты содержат такие ионы.
Введение в коллоидные растворы индифферентных электролитов сопровождается двумя явлениями: 1) ионным обменом между противоионами диффузного слоя ДЭС и ионами добавленного электролита; 2) сжатием диффузной атмосферы вокруг поверхности частиц.
Концентрационная коагуляция наблюдается в золях с высоким j-потенциалом частиц при увеличении концентрации электролита, т.е. ионной силы раствора. Этот механизм коагуляции осуществляется при действии индифферентных электролитов, не способных к избирательной адсорбции. Добавление таких электролитов не изменяет величину j-потенциала во внутренней обкладке двойного слоя. В этом случае коагуляцию вызывают электростатический эффект сжатия двойного электрического слоя и связанное с ним уменьшение z-потенциала
Сжатие диффузионного слоя является следствием двух причин:
1) перемещения части противоионов из диффузионного слоя в адсорбционный слой, что ведет к дополнительной компенсации z-потенциала;
2) подавления диффузии противоионов и уменьшения размытости диффузного слоя за счет увеличения ионной силы дисперсионной среды. Этот фактор является преобладающим для систем с сильно заряженными частицами.
Рассмотрим влияние индифферентных электролитов NaCl, Na2SO4, K3[Fe(CN)6], содержащих одно-, двух- и трехзарядные ионы-коагуляторы на ϕ3m+- и ζ3х+-потенциалы ДЭС золя гидроксида железа.
Влияние электролита NaCl на ϕ3+- и ζ3х+-потенциалы ДЭС золя Fe(OH)3
К агрегативно устойчивому золю Fe(OH)3 с положительно заряженными коллоидными частицами добавляем коагулирующий электролит NaCl.
По отношению к исходному золю вводимый электролит NaCl будет индифферентным, так как в нём отсутствуют ионы, способные к избирательной адсорбции. По правилу Шульце-Гарди ионами-коагуляторами во вводимом электролите NaCl будут ионы Cl– , т.к. их заряд противоположен заряду коллоидных частиц. При введении электролита в золь ионы-коагуляторы Cl– за счет электростатических сил притяжения входят в диффузную часть ДЭС, вытесняя при этом эквивалентное количество противоионов диффузного слоя Cl– в объём дисперсионной среды (воды). При ионном обмене однозарядных ионов-коагуляторов Cl– на однозарядные противоионы диффузного слоя Cl– ,
количество противоионов в диффузном слое не меняется, следовательно, не меняется величина ζ3x+ потенциала (ζ3x+ = const), т.к. она зависит от количества противоионов в диффузном слое, при этом величина φ3m+ = const, т.к. она зависит от количества адсорбированных ПОИ (m).
При увеличении концентрации электролита начинается сжатие диффузной части ДЭС и переход противоионов из диффузного слоя в адсорбированный в результате увеличения ионной силы раствора. При переходе количество противоионов в диффузном слое уменьшается, следовательно, уменьшается величина ζ3x+, при этом величина φ3m+ = const.
При увеличении концентрации электролита наблюдается дальнейшее уменьшении величины ζ3x+, при этом величина φ3m+ = const
(рис. 2, участок аб).
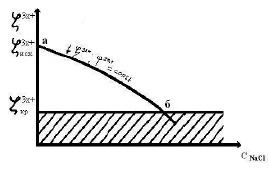
Рис. 2. Зависимость ζ3x+ золя от концентрации электролита NaCl
В точке «б» величина ζ3m+ – потенциала достигает величины ζ3x+ критическая и золь коагулирует по концентрационному механизму.
Влияние электролита Na2SO4 на j3+- и ζ3х+-потенциалы ДЭС золя Fe(OH)3
К агрегативно устойчивому золю Fe(OH)3 с положительно заряженными коллоидными частицами добавляем коагулирующий электролит Na2SO4.

По отношению к исходному золю вводимый электролит будет индифферентным, так как в нём отсутствуют ионы, способные к избирательной адсорбции. По правилу Шульце-Гарди ионами-коагуляторами во вводимом электролите будут SO42–, так как их заряд противоположен заряду коллоидных частиц. При введении электролита в золь ионы-коагуляторы SO42– за счет электростатических сил притяжения входят в диффузную часть ДЭС, вытесняя при этом эквивалентное количество противоионов диффузного слоя Cl– в объём дисперсионной среды (воды). При ионном обмене двух зарядных ионов-коагуляторов SO42– на однозарядные противоионы диффузного Cl-, количество противоионов в диффузном слое уменьшается, следовательно, уменьшается величина ζ3x+-потенциала, потому что величина ζ3x+-потенциала зависит от количества противоионов в диффузном слое; при этом φ3m+ остаётся постоянным, так как величина φ3m+-потенциала зависит от количества адсорбционных ПОИ. При ионном обмене сжатие диффузной части ДЭС будет происходить намного быстрее по сравнению с однозарядными ионами-коагуляторами. Также при ионном обмене диффузный слой будет содержать смесь ионов Cl- и SO42–. При дальнейшем увеличении концентрации Na2SO4 начинается переход противоионов из диффузного слоя в адсорбционный в результате увеличения ионной силы раствора. При переходе количество противоионов в диффузном слое уменьшается, следовательно, уменьшается величина ζ3x+; φ3m+ = const.
При дальнейшем увеличении концентрации Na2SO4 наблюдается дальнейшее уменьшение величины ζ3x+; при этом φ3m+=const (рис. 3, участок аб).
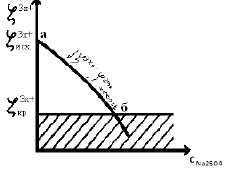
Рис. 3. Зависимость ζ3x+ золя от концентрации электролита Na2SO4
В точке «б» величина ζ3x+-потенциала достигает величины ζ3x+ критическое и золь коагулирует по концентрационному механизму.
Влияние электролита K3[Fe(CN)6] на ϕ3+- и ζ3х+-потенциалы ДЭС золя Fe(OH)3
К агрегативно устойчивому золю Fe(OH)3 с положительно заряженными коллоидными частицами добавляем коагулирующий электролит K3[Fe(CN)6].

По отношению к исходному золю Fe(OH)3 электролит K3[Fe(CN)6] является индифферентным, так как в нём отсутствуют ионы, способные к избирательной адсорбции. По правилу Шульце-Гарди ионами-коагуляторами будут ионы [Fe(CN)]3–, так как их заряд противоположен заряду коллоидных частиц. При введении электролита в золь ионы-коагуляторы [Fe(CN)]3– за счет электростатических сил притяжения входят в диффузную часть ДЭС, вытесняя при этом эквивалентное количество противоионов диффузного слоя Cl– в объём дисперсионной среды (воды). При ионном обмене трехзарядных ионов-коагуляторов [Fe(CN)]3– на однозарядные противоионы диффузного слоя Cl-, количество противоионов в диффузном слое уменьшается, следовательно, уменьшается величина ζ3x+, так как величина ζ3x+ потенциала зависит от количества противоионов в диффузном слое; при этом φ3m+ = const, так как величина φ3m+-потенциала зависит от количества адсорбируемых ПОИ. При ионном обмене сжатие диффузной части ДЭС будет происходить намного быстрее по сравнению с одно- и двухзарядными ионами-коагуляторами. Также при ионном обмене диффузный слой будет содержать смесь ионов Cl– и [Fe(CN)]3–. При увеличении концентрации электролита K3[Fe(CN)6] начинается переход противоионов из диффузного слоя в адсорбированный в результате увеличения ионной силы раствора. При переходе количество противоионов в диффузном слое уменьшается, следовательно, уменьшается величина ζ3x+; при этом φ3m+ = const. При дальнейшем увеличении концентрации электролита K3[Fe(CN)6] наблюдается дальнейшее уменьшение величины ζ2x+; при этом φ3m+=const (рис. 4, участок аб).
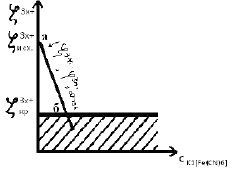
Рис. 4. Зависимость ζ3x+ золя от концентрации электролита K3[Fe(CN)6]
В точке «б» величина ζ3x+-потенциала достигает величины ζ3x+ критическое и золь коагулирует по концентрационному механизму.
Заключение
Проведенные исследования подтвердили закономерности найденные Шульце-Гарди: коагулирующим действием обладает тот ион электролита заряд, которого противоположен заряду коллоидных частиц; коагулирующее действие тем сильнее, чем выше заряд иона коагулятора.
На основании минимального объема электролитов, вызвавшего коагуляцию золя Fe(OH)3, рассчитанных величин порогов коагуляции электролитов можно сделать вывод, что лучшим коагулятором для золя Fe(OH)3 будет являться электролит K3[Fe(CN)6], а худшим – NaCl.
Изучение механизма коагуляции золя Fe(OH)3 индифферентными электролитами NaCl, Na2SO4, K3[Fe(CN)6] показало, что коагуляция протекает по концентрационному механизму, который сопровождается следующими явлениями: ионным обменом между вводимыми ионами-коагуляторами и противоионами диффузного слоя, сжатием диффузной части ДЭС и переходом противоинов из диффузного слоя в адсорбционный в результате увеличения ионной силы раствора.
Библиографическая ссылка
Васильев С.Е. КОАГУЛЯЦИЯ ЗОЛЯ ГИДРОКСИДА ЖЕЛЕЗА (III) ПОД ДЕЙСТВИЕМ ЭЛЕКТРОЛИТОВ. МЕХАНИЗМ КОАГУЛЯЦИИ // Международный школьный научный вестник. 2019. № 2-3. ;URL: https://school-herald.ru/ru/article/view?id=989 (дата обращения: 12.05.2026).